
A pair of heart devices linked to hundreds of injuries and at least 14 deaths has received the FDA’s most serious recall, the agency .
The recall comes years after surgeons say they first noticed problems with the HeartMate II and HeartMate 3, manufactured by Thoratec Corp., a subsidiary of Abbott Laboratories. The devices are not currently being removed from the market. In an emailed response, Abbott said it had communicated the risk to customers this year.
The delayed action raises questions for some safety advocates about how and when issues with approved medical devices should be reported. The heart devices in question have been associated with thousands of reports of patients‚Äô injuries and deaths, as described in a ƒ¢πΩ”∞‘∫ Health News investigation late last year.
“Why doesn’t the public know?” said , a cardiologist and an expert in medical device safety and regulation at the University of California-San Francisco. Though some surgeons may have been aware of issues, others, particularly those who do not implant the device frequently, may have been in the dark. “And their patients are suffering adverse events,” he said.
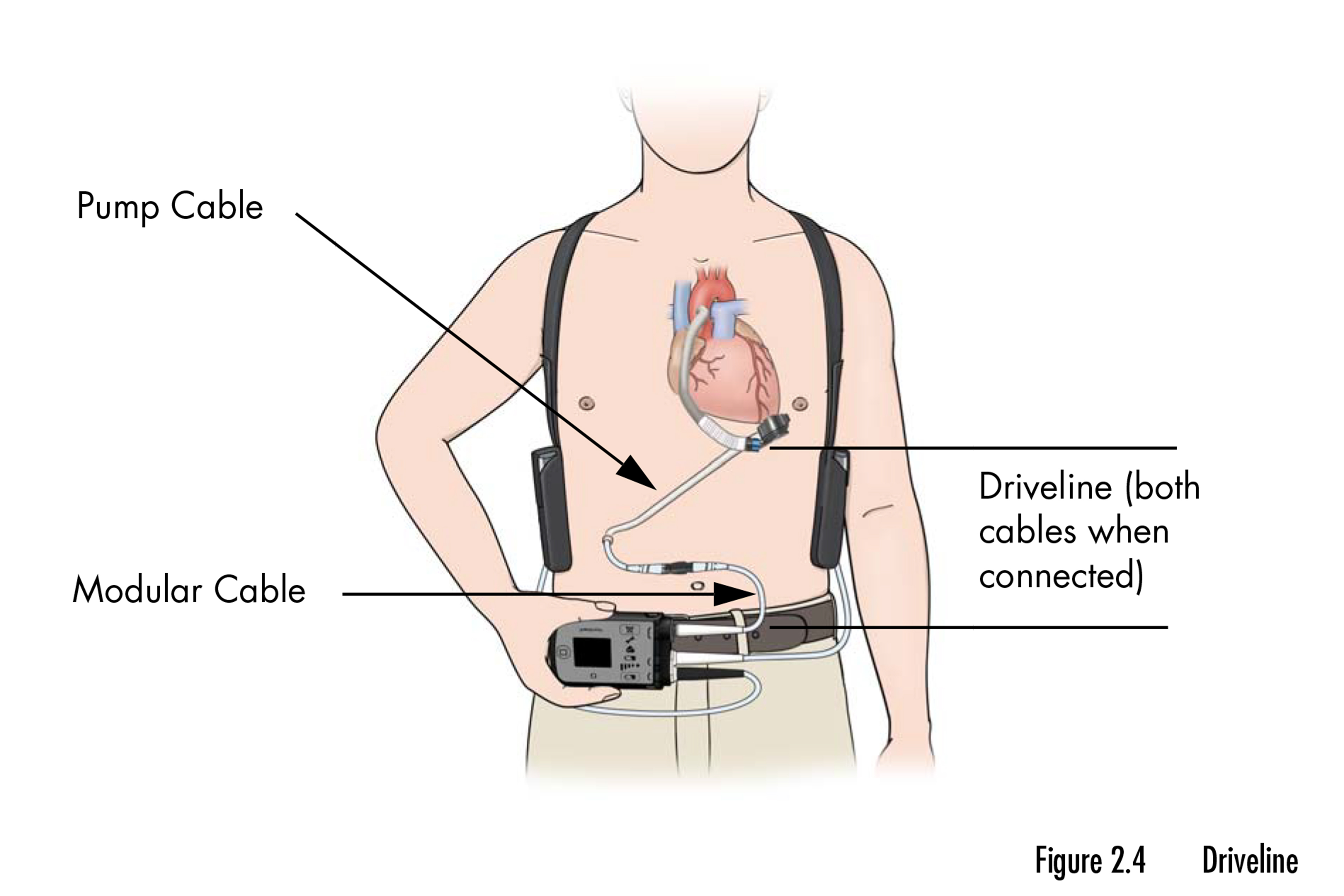
The recall involves a pair of mechanical pumps that help the heart pump blood when it can’t do so on its own. The devices, small enough to fit in the palm of a hand, are implanted in patients with end-stage heart failure who are waiting for a transplant or as a permanent solution when a transplant is not an option. The recall affects nearly 14,000 devices.
Amanda Hils, an FDA press officer, said the agency is working with Abbott to investigate the reported injuries and deaths and determine if further action is needed.
“To date, the number of deaths reported appears consistent with the ,” Hils said in an email.
According to the FDA’s recall notice, the devices can cause buildup of “biological material” that reduces their ability to help the heart circulate blood and keep patients alive. The buildup accumulates gradually and can appear two years or more after a device is implanted in a patient’s chest.
Doctors were advised to watch out for “low-flow alarms” on the devices and, if they do diagnose the obstruction, to either monitor the patient or perform surgery to implant a stent, release the blockage, or replace the pump. ‚ÄúRates of outflow obstruction are low,‚Äù Abbott spokesperson Justin Paquette said in an email, adding that patients whose devices are functioning normally ‚Äúhave no reason for concern.‚Äù
A review of the FDA device database shows at least 130 reports related to HeartMate II or 3 that mention the complication reported by regulators. The earliest such report filed with the FDA dates to at least 2020, according to a ƒ¢πΩ”∞‘∫ Health News review of the database.
Monday’s alert is the second Class 1 recall of a HeartMate device this year.
In January, Abbott issued an urgent ‚Äú‚Äù to hospitals about in which the HeartMate 3 unintentionally starts and stops due to the pump’s communication system, which cardiologists use to assess patients’ status. The FDA in March.
In February, Abbott issued to hospitals about the blockage problem, asking them to inform physicians, complete and return an acknowledgment form, and pay attention to low-flow alarms on the device’s monitor that may indicate an obstruction. The company said in the letter that it is working on “a design solution” to prevent the blockages.
A in the Journal of Thoracic and Cardiovascular Surgery reported the obstruction in about 3% of cases, though the incidence rate was higher the longer a patient had the device.
The only other Class 1 was in May 2018, when the company issued corrective action notices to hospitals and physicians warning that the graft line that carries blood from the pump to the aorta could twist and stop blood flow.
The FDA recall notice issued Monday includes to diagnose the blockage using an algorithm to detect obstructions and, if needed, a CT angiogram to verify the cause.
At present, the HeartMate 3, which was first approved by the FDA in 2017, is the only medical option for many patients with end-stage heart failure and who do not qualify for a transplant. The HeartMate 3 has supplanted the HeartMate II, which received FDA approval in 2008.
If the new recall leads to the device being removed from the market, end-stage heart failure patients could have no options, said , a cardiothoracic surgeon at the University of Michigan who also oversees a proprietary database of HeartMate II and HeartMate 3 implants.
If that happens, “we are in trouble,” Pagani said. “It would be devastating to the patients to not have this option. It’s not a perfect option — no pump ever is — but this is as good as it’s ever been.”
It’s not known precisely how many patients have received a HeartMate II or HeartMate 3 implant. That information is proprietary. The FDA recall notices show worldwide distribution of more than and more than .
The blockage complication may have gone unreported to the public for so long partly because physicians are not required to report adverse events to federal regulators, said Madris Kinard, a former FDA medical device official and founder of , a company that makes FDA device data more user-friendly for hospitals, law firms, and investors.
Only device manufacturers, device importers, and hospitals are to report device-related injuries, deaths, and significant malfunctions to the FDA.
“If this is something physicians were aware of, but they weren’t mandated to report to the FDA,” Kinard said, “at what point does that communication between those two groups need to happen?”
Dhruva, the cardiologist, said he is looking for transparency from Abbott about what the company is doing to address the problem so he can have more thorough conversations with patients considering a HeartMate device.
‚ÄúWe‚Äôre going to expect to have some data saying, ‚ÄòHey we created this fix, and this fix works, and it doesn‚Äôt cause a new problem.‚Äô That‚Äôs what I want to know,‚Äù he said. ‚ÄúThere‚Äôs just a ton more that I feel in the dark about, to be honest, and I’m sure that patients and their families do as well.‚Äù
[Update: This article was updated at 5:20 p.m. ET on April 16, 2024, with a response from Abbott Laboratories, which it provided after publication.]



